


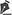
Case Report - Year 2018 - Volume 33 -
Relato de caso: síndrome de Klippel-Trénaunay-Weber
Case Report: Klippel-Trénaunay-Weber syndrome
RESUMO
Introdução: A síndrome de Klippel-Trénaunay-Weber (SKTW) é caracterizada pelo conjunto de
sinais que consiste em malformações capilares, malformações venosas com ou
sem malformações linfáticas associado ao supercrescimento de membros. Na
maioria das vezes, envolve apenas uma extremidade com malformação
arteriovenosa e cerca de 75% dos pacientes manifestam antes dos 10 anos de
idade.
Relato de Caso: Relatamos um caso de Klippel-Trénaunay-Weber em um paciente de 7 meses em
acompanhamento na enfermaria da Cirurgia Plástica do Hospital de Clínicas da
Universidade Federal de Uberlândia para o qual foi proposto tratamento
cirúrgico da lesão.
Conclusão: Como a SKTW é uma doença com morbidade progressiva e grave, o paciente deve
ser acompanhado em um centro de referência com experiência e arsenal
terapêutico diversificado para atuar da melhor forma possível no
tratamento.
Palavras-chave: Síndrome de Klippel-Trenaunay-Weber; Malformações vasculares; Hemangioma
ABSTRACT
Introduction: The Klippel-Trénaunay-Weber syndrome
(KTWS) is characterized by several signs, including capillary
malformations and venous malformations with or without
lymphatic malformations associated with limb overgrowth. In
most cases, only one extremity is involved with arteriovenous
malformation, and approximately 75% of the patients manifest
symptoms before 10 years of age.
Case Report: We report a case
of a 7-month-old patient with KTWS followed-up at the Plastic
Surgery Service of the Hospital de Clínicas, Federal University of
Uberlândia; surgical treatment of the lesion was proposed for the
patient.
Conclusion: Since KTWS is a progressive disease with
severe morbidity, the patient must be followed-up at a reference
center by experienced staff with diverse therapeutic arsenal.
Keywords: Klippel-Trénaunay-Weber syndrome; Vascular malformations; Hemangioma
INTRODUÇÃO
A síndrome de Klippel-Trenaunay-Weber (SKTW) é caracterizada pelo conjunto de sinais que consiste em malformações capilares, malformações venosas com ou sem malformações linfáticas associado ao supercrescimento de membros1. Na maioria das vezes, envolve apenas uma extremidade com malformação arteriovenosa e cerca de 75% dos pacientes manifestam a afecção antes dos 10 anos de idade2,3.
A síndrome de Klippel-Trenaunay foi descrita pela primeira vez por Maurice Klippel e Paul Trenaunay. Eles relataram dois casos que possuíam a tríade (mancha em vinho do porto, varizes e hipertrofia óssea e de tecidos moles) em comum4. Algum tempo depois, Frederick Weber descreveu alguns casos apresentando semelhança com a tríade, tendo a presença de fístula arteriovenosa como associação3,4. A SKTW e a Sindrome de Parkes Weber podem ser consideradas em conjunto como sd de Klippel-Trenaunay-Weber por apresentar sinais clínicos diferentes de uma única enfermidade5,6.
A síndrome de Klippel-Trenaunay-Weber é um distúrbio mesodérmico congênito raro, sendo registrados cerca de mil casos em todo o mundo7. A SKTW distribui-se igualmente entre os diversos grupos étnicos e afeta mais homens, na proporção de 1,5:17,8. A etiologia da síndrome permanece desconhecida, embora existam algumas teorias de sua patogenia9.
Embora a SKTW seja uma condição esporádica, trabalhos relatam casos familiares de SKTW que não foram herdados de um padrão mendeliano, sugerindo uma herança multifatorial, com penetração e expressão variáveis. Estudos realizados posteriormente por Happle sugerem que a herança de um único gene defeituoso adquirido na embriogênese poderia explicar o desenvolvimento dessa síndrome, bem como a ocorrência de casos esporádicos e familiares, sugerindo que a herança autossômica dominante é mais provável10,11.
Clinicamente, a SKTW compreende hemangioma plano, alterações venosas como má-formações e varizes, hipertrofia óssea e de tecidos moles12,13.
O diagnóstico é clínico e pode ser feito pela presença da tríade de anormalidades, ou sendo necessários apenas dois sinais da tríade. Geralmente os pacientes cursam com manchas em vinho do porto, desde o nascimento, principalmente em membro hipertrofiado, variando em profundidade14. As má- formações venosas acometem os membros inferiores na grande maioria dos casos. Angiodisplasias arteriais ou venosas podem estar presentes em qualquer região do corpo, desde a pele até órgãos viscerais. Por isso, existe a possibilidade ocorrer flebite, sangramento, trombose venosa profunda (17% dos pacientes), embolia pulmonar, hemoperitônio, hemotórax e insuficiência venosa crônica15.
Trata-se de uma síndrome pouco frequente em nosso meio, porém merece destaque pelo fato da progressiva e grave morbidade.
RELATO DO CASO
D.A.P., 7 meses, masculino, natural e procedente de Anápolis-GO, encaminhado ao nosso serviço do Pronto Socorro do Hospital de Clínicas da Universidade Federal de Uberlândia, MG, em 29/12/2014 com quadro de febre, vômitos, diarreia e desidratação havia 2 dias. Associado a esse quadro, apresentava hemangioma gigante em membro inferior esquerdo, varizes e hipertrofia óssea e de tecidos moles (síndrome de Klippel-Trenaunay-Weber) (Figura 1) com antecedente de tratamento prévio recente a laser em outro serviço de Goiânia-GO e em uso de propranolol.
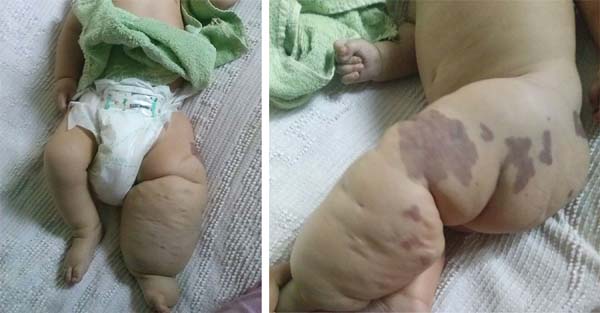

Evoluiu-se rapidamente e gravemente com necrose do membro inferior esquerdo (MIE) até a região glútea ipsilateral e com quadro de choque séptico refratário (Figura 2). Recebeu tratamento intensivo em CTI com drogas vasopressoras e antibioticoterapia de amplo espectro por 28 dias. Na avaliação da cirurgia vascular, foi realizado o ecoDoppler arterial e venoso, constatando-se a presença de malformação e pequenas fístulas arteriovenosa da extremidade esquerda, confirmando o diagnóstico síndrome de Klippel-Trenaunay-Weber. Contudo, permaneceu com lesão necrótica extensa em MIE sendo necessário, primariamente, desbridamento cirúrgico do MIE e posteriormente desarticulação coxofemoral esquerda com colostomia protetora em 28/01/2015.
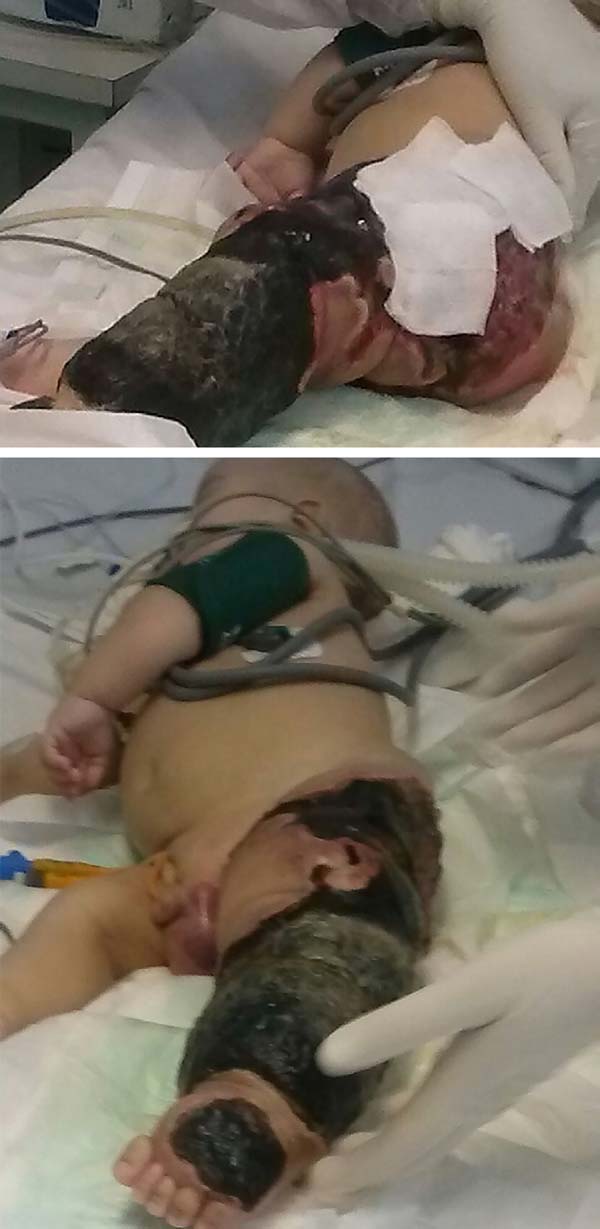
Após 2 meses de tratamento clínico intensivo, houve melhora clínica e tópica da lesão em região de pós-operatório da desarticulação (Figura 3A), sendo realizada enxertia de pele em malha com uso do dermátomo elétrico e do Mash Graft para oclusão da ferida cruenta (Figura 3B), sendo a área doadora de coxa direita. O paciente apresentou boa recuperação clínica e epitelização da área receptora e doadora, obtendo alta hospitalar no 15º dia de pós-operatório (Figura 3C).
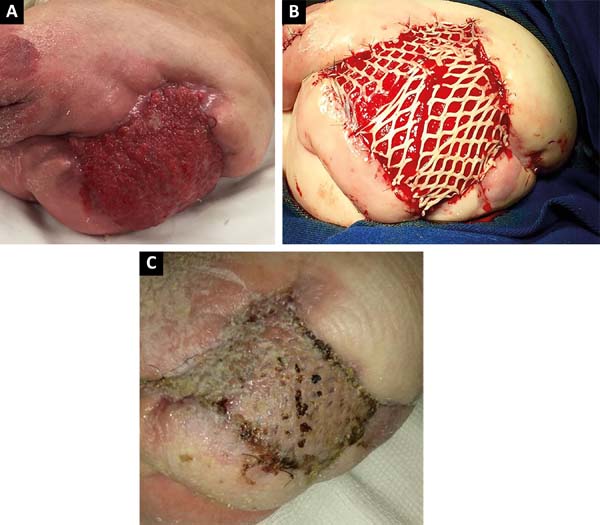

O paciente está em acompanhamento ambulatorial neste serviço, evoluindo satisfatoriamente com epitelização completa da área doadora e receptora. Já foi realizado fechamento da colostomia de proteção com a reconstrução do trânsito intestinal.
DISCUSSÃO
Este estudo foi motivado por ter recebido um paciente referindo tratamento prévio em outro serviço de Cirurgia Plástica com tratamento conservador da SKTW com laser. Tal conduta manteve o paciente incapacitado, sofrendo com sintomas de estase venoso crônico e evoluiu com complicações de infecção locais de pele até receber o tratamento final.
Não existe nenhum tratamento curativo, e os objetivos terapêuticos são destinados a melhorar os sintomas do paciente e corrigir as consequências de lesões graves e a discrepância de comprimento. No entanto, todos os autores concordam que as medidas conservadoras continuam norteando o tratamento da SKTW. Isso não afasta a necessidade de intervenções cirúrgicas pontuais durante a evolução da história natural da doença.
As terapias adjuvantes podem variar desde a terapia a laser, escleroterapia com microespuma, ressecções escalonadas de veias ectasiadas e até exéreses mais amplas1,16-19. As indicações mais utilizadas para o tratamento operatório são: as hemorragias, as infecções locais, o tromboembolismo e a ocorrência de ulcerações de perna muito refratárias. Outras indicações são: dor local, limitação funcional e estética17.
A radioterapia intervencionista tem grande papel na propedêutica das malformações arteriovenosas (MAVs). Por meio da mesma, é possível avaliar o tipo de malformação e como os vasos de alimentação são estruturados. Para o tratamento de MAV de baixo fluxo (STWK), em alguns casos pode ser aplicada uma injeção de agente esclerosante para tornar os vasos menores.
Em outros, isso pode ser feito sob fluoroscopia. Há opções limitadas de tratamento para displasia venosa congênitas. Em casos graves, o intervencionista pode usar remoção cirúrgica, escleroterapia ou uma técnica de ablação endovascular. Se houver sintomas na pele, tais como uma mancha vinho do porto, poderá ser indicado o tratamento a laser.
Em casos como o supracitado, considerando o tratamento realizado, a cobertura da área cruenta pela enxertia pode ser vista como uma boa opção reconstrutiva, visto a simplicidade do procedimento, menor morbidade quando comparamos com o uso dos retalhos e possibilidade de reabilitação através do uso de próteses.
Apesar de ser o mais alto nível de amputação em membro inferior, a protetização é eficiente, de vez que a prótese para este nível de amputação proporciona segurança e estabilidade, com marcha contínua, com ou sem auxiliar de locomoção, dependendo de outros fatores, entre eles a idade do paciente.
A SKTW deve ser suspeitada em todos os recém-nascidos com malformações capilares envolvendo uma extremidade do corpo desde o nascimento. O diagnóstico diferencial para a SKTW é a síndrome de Proteus e a síndrome de Maffucci, entre outras malformações capilares da pele não síndrômicas20.
Avanços maiores serão feitos quando for possível diagnosticar ainda mais precocemente a SKTW e impedir o desenvolvimento da hipertrofia de tecidos, angiodisplasias complexas e outras alterações fenotípicas, talvez corrigindo ou impedindo a mutação genética relacionada20.
CONCLUSÃO
A SKTW é uma doença rara, com morbidade progressiva e grave. O paciente portador da síndrome deve ser acompanhado em um centro de referência com experiência e arsenal terapêutico diversificado para atuar da melhor forma possível no tratamento. Cada paciente tem parcimônia e individualização na escolha do melhor tratamento, bem como a época ideal de realizá-lo.
Atualmente, a indicação de intervenções operatórias é restrita às complicações decorrentes do quadro inicial. No presente caso, a utilização do tratamento operatório foi decisivo para permitir uma melhora no quadro clínico e de qualidade de vida do paciente, mostrando que pode ser uma alternativa válida.
REFERENCES
1. Villela ALC, Guedes LGS, Paschoa VVA, David AB, Tenório TM, Lamego Junior HP, et al. Perfil epidemiológico de 58 portadores de síndrome de Klippel-Trénaunay-Weber acompanhados no Ambulatório da Santa Casa de São Paulo. J Vasc Bras. 2009;8(3):219-24. DOI: http://dx.doi.org/10.1590/S1677-54492009000300006
2. Favorito LA. Vesical Hemangioma in patient with Klippel-Trenaunay-Weber syndrome. J Urol. 2003;29(2):149-50.
3. Tonsgard JH, Fasullo M, Windle ML, McGovern M, Petry PD, Buehler B. Klippel-Trenaunay-Weber Syndrome [Internet]. Pediatrics: General Medicine Articles 2006. Disponível em: http://www.emedicine.com/derm/topic213.htm
4. Klippel M, Trénaunay P. Du naevus variqueux osteohypertrophique. Arch Gen Med (Paris). 1900;185:641-72.
5. Weber FP. Haemangiectatic hypertrophy of limbs: congenital phlebarteriectasis and so-called congenital varicose veins. Br J Child Dis. 1918;15:13-7.
6. Kihiczak GG, Meine JG, Schwartz RA, Janniger CK. Klippel-Trenaunay syndrome: a multisystem disorder possibly resulting from a pathogenic gene for vascular and tissue overgrowth. Int J Dermatol. 2006;45(8):883-90. PMID: 16911369 DOI: http://dx.doi.org/10.1111/j.1365-4632.2006.02940.x
7. Berry SA, Peterson C, Mize W, Bloom K, Zachary C, Blasco P, et al. Klippel-Trenaunay syndrome. Am J Med Genet. 1998;79(4):319-26. PMID: 9781914 DOI: http://dx.doi.org/10.1002/(SICI)1096-8628(19981002)79:4<319::AID-AJMG15>3.0.CO;2-U
8. Capraro PA, Fisher J, Hammond DC, Grossman JA. Klippel-Trenaunay syndrome. Plast Reconstr Surg. 2002;109(6):2052-60; quiz 2061-2. DOI: http://dx.doi.org/10.1097/00006534-200205000-00041
9. Meine JG, Schwartz RA, Janniger CK. Klippel-Trenaunay-Weber syndrome. Cutis. 1997;60(3):127-32. PMID: 9314616
10. Lorda-Sanchez I, Prieto L, Rodriguez-Pinilla E, Martinez-Frias ML. Increased parental age and number of pregnancies in Klippel-Trenaunay-Weber syndrome. Ann Hum Genet. 1998;62(Pt 3):235-9. DOI: http://dx.doi.org/10.1046/j.1469-1809.1998.6230235.x
11. Hergesell K, Kröger K, Petruschkat S, Santosa F, Herborn C, Rudofsky G. Klippel-Trenaunay syndrome and pregnancy. Int Angiol. 2003;22(2):194-8.
12. Wang QK. Update on the molecular genetics of vascular anomalies. Lymphat Res Biol. 2005;3(4):226-33. DOI: http://dx.doi.org/10.1089/lrb.2005.3.226
13. Delis KT, Gloviczki P, Wennberg PW, Rooke TW, Driscoll DJ. Hemodynamic impairment, venous segmental disease, and clinical severity scoring in limbs with Klippel-Trenaunay syndrome. J Vasc Surg. 2007;45(3):561-7. PMID: 17275246 DOI: http://dx.doi.org/10.1016/j.jvs.2006.11.032
14. Viljoen D, Saxe N, Pearn J, Beighton P. The cutaneous manifestations of the Klippel-Trenaunay-Weber syndrome. Clin Exp Dermatol. 1987;12(1):12-7. DOI: http://dx.doi.org/10.1111/j.1365-2230.1987.tb01845.x
15. Kihiczak GG, Meine JG, Schwartz RA, Janniger CK. Klippel-Trenaunay syndrome: a multisystem disorder possibly resulting from a pathogenic gene for vascular and tissue overgrowth. Int J Dermatol. 2006;45(8):883-90. PMID: 16911369 DOI: http://dx.doi.org/10.1111/j.1365-4632.2006.02940.x
16. Lindenauer SM. The Klippel-Trenaunay Syndrome: Varicosity, Hypertrophy and Hemangioma With No Arteriovenous Fistula. Ann Surg. 1965;162(2):303-4. DOI: http://dx.doi.org/10.1097/00000658-196508000-00023
17. Gloviczki P, Driscoll DJ. Klippel-Trenaunay syndrome: current management. Phlebology. 2007;22(6):291-8. DOI: http://dx.doi.org/10.1258/026835507782655209
18. Frasier K, Giangola G, Rosen R, Ginat DT. Endovascular radiofrequency ablation: a novel treatment of venous insufficiency in Klippel-Trenaunay patients. J Vasc Surg. 2008;47(6):1339-45. DOI: http://dx.doi.org/10.1016/j.jvs.2008.01.040
19. Delis KT, Gloviczki P, Wennberg PW, Rooke TW, Driscoll DJ. Hemodynamic impairment, venous segmental disease, and clinical severity scoring in limbs with Klippel-Trenaunay syndrome. J Vasc Surg. 2007;45(3):561-7. PMID: 17275246 DOI: http://dx.doi.org/10.1016/j.jvs.2006.11.032
20. Garzon MC, Huang JT, Enjolras O, Frieden IJ. Vascular malformations. Part II: associated syndromes. J Am Acad Dermatol. 2007;56(4):541-64. PMID: 17367610
1. Universidade Federal de Uberlândia, Uberlândia,
MG, Brasil.
Autor correspondente: Victor Parreira Bizinoto, Av. Mato Grosso, nº 3395, apt. 202 - Umuarama - Uberlândia, MG, Brasil. CEP 38405-314. E-mail: victorpbizinoto@hotmail.com
Artigo submetido: 26/11/2017.
Artigo aceito: 5/9/2018.
Conflitos de interesse: não há.
 Read in Portuguese
Read in Portuguese
 Read in English
Read in English
 PDF PT
PDF PT
 Print
Print
 Send this article by email
Send this article by email
 How to Cite
How to Cite
 Mendeley
Mendeley
 Pocket
Pocket