ISSN Online: 2177-1235 | ISSN Print: 1983-5175
Neurofibromatose: relato de caso
Membro Superior e Inferior -
Year2018 -
Volume33 -
(Suppl.1)
Inara do Carmo Lucchese; Diego Fernando Villagra Avila; Estevão José Muller Uliano; Leandro Soares Grangeiro; Zulmar Antonio Accioli de Vasconcellos; Jorge Bins Ely
http://www.dx.doi.org/10.5935/2177-1235.2018RBCP0071
RESUMO
Os autores relatam um caso de neurofibromatose em um paciente que desenvolveu uma volumosa tumoração lombar, alertando para as variações clínicas desta afecção.
Palavras-chave:
Neurofibromatoses; Neurofibroma; Neoplasias.
INTRODUÇÃO
Neurofibromatose é numa doença autossômica dominante com penetrância irregular e de expressividade variável1. Pode ser classificada em neurofibromatose tipo 1 (NF1), neurofibromatose tipo 2 (NF2) e schwannomatose1.
A NF1 ocorre em 1 para cada 2.500 a 3.000 nascidos vivos, de todas as raças2. Pode ser caracterizada por manchas "café-com-leite", efélides (sardas), neurofibromas da pele, nódulos na íris de Lisch, displasia de ossos longos, glioma do nervo óptico e neurofibroma plexiforme3. É causada por mutações no cromossomo 17, que resultam em disfunção de uma proteína supressora de tumores denominada neurofibrina. É uma doença herdada de um dos pais em 50% dos casos. Os demais não apresentam história familiar, sugerindo novas mutações2.
A , ou central, é mais rara e resulta de mutações no cromossomo 22, levando à disfunção da proteína merlina, provocando crescimento de tumores no sistema nervoso4.
Já a schwannomatose, apresenta localização genética e defeitos moleculares ainda não bem estabelecidos e tem como principal característica a dor neuropática intratável5.
O diagnóstico da neurofibromatose é baseado em achados clínicos, sendo que pode ser realizada biópsia de um nódulo subcutâneo6.
O tratamento limita-se a excisão dos tumores que produzem sintomas ou alterações funcionais ou estéticas ao paciente6.
OBJETIVO
Relato de caso de neurofibroma.
RELATO DE CASO
Paciente masculino, 26 anos, procurou atendimento no Serviço de Cirurgia Plástica do Hospital Universitário com queixa de tumoração volumosa em região lombar. História pregressa de neurofibromatose tipo 1. A lesão apresentava 12 anos de evolução, com crescimento progressivo, porém assintomática.
Ao exame físico, identificou-se lesão localizada em região posterolateral do abdômen à direita, cerca de 20cm x 20cm x 10cm, de consistência firme, fixa à pele, indolor à palpação (Figuras 1 e 2). Cicatriz prévia de cirurgia por escoliose em dorso (mediana e à esquerda).
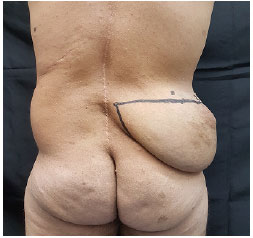 Figura 1.
Figura 1. Visão posterior.
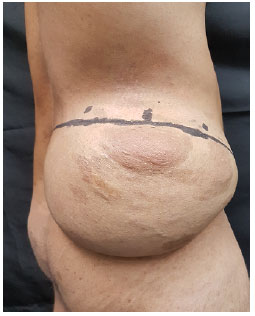 Figura 2.
Figura 2. Visão Lateral.
Realizada tomografia que descreve massa de tecido adiposo na topografia referida, delimitada pela fáscia toracolombar associada a espessamento das camadas mais superficiais da pele e áreas de densificação linear podendo corresponder a vasos, e não descartando possibilidade de tumor de linhagem adiposa.
Paciente foi submetido à cirurgia, sob anestesia geral, com vistas à ressecção tumoral. Realizado incisão na margem inferior da lesão, identificada massa hipervascularizada sem, no entanto, identificação de plano de clivagem ou cápsula (Figura 3). Ressecada toda a lesão, que pesou 1.300 Kg. Paciente recebeu alta no dia seguinte, com dreno de portovac. Paciente apresentou boa evolução no pós-operatório (Figura 4). O exame anatomopatológico concluiu ser uma neoplasia neurofibromatosa de padrão plexiforme com atipias. Embora não haja critérios de malignidade, o paciente necessitará seguimento devido à impossibilidade de definir o comportamento biológico da lesão.
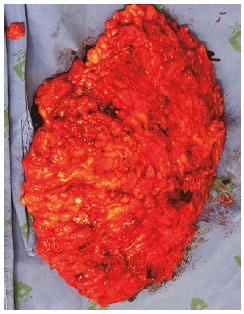 Figura 3.
Figura 3. Peça ressecada.
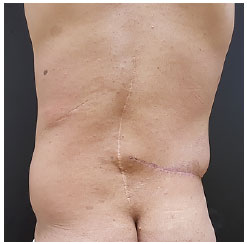 Figura 4.
Figura 4. Pós-operatório (cicatriz transversa à direita).
DISCUSSÃO
O primeiro relato de neurofibromatose data de 1768, com a descrição de neurofibromas cutâneos. Em 1882, o médico alemão Von Recklinghausen descreveu a afecção de maneira completa, confirmando a origem nervosa dos tumores. Até hoje, a doença também é conhecida por seu nome1.
A neurofibromatose é classificada em 3 tipos, sendo a NF1, NF2 e schwannomatose.
A NF1, mais comum, ocorre em 1 para cada 2.500 a 3.000 nascidos vivos, de qualquer raça2. É uma doença genética autossômica dominante, causada por mutações no cromossomo 17, que resultam em disfunção de uma proteína supressora de tumores denominada neurofibrina2. As principais características clínicas da NF1 são as manchas "café-com-leite", os neurofibromas dérmicos e plexiforme, as efélides axilares e/ou inguinais e os nódulos de Lisch7.
Trata-se de uma afecção multissistêmica com possibilidade de acometimento oftalmológico, osteomuscular, cardiovascular, endócrino, do sistema nervoso central e periférico e da aprendizagem8. Os tumores cutâneos podem assumir formas sésseis, pedunculadas, cônicas ou lobuladas. Podem ser discretas e pequenas ou atingir grandes proporções, sendo então referida como "le tumeur royale9". Outra característica das lesões é não serem encapsuladas e não apresentarem um plano de clivagem.
No caso descrito, o paciente apresentava essa forma de acometimento cutâneo, sem outras alterações, mas acarretando grande transtorno psicológico e estético.
A NF2, mais rara, resulta de mutações no cromossomo 22, levando à disfunção da proteína merlina, provocando crescimento de tumores no sistema nervoso4.
A schwannomatose, também rara, apresenta localização genética e defeito moleculares ainda não bem estabelecidos e tem como principal característica a dor neuropática intratável5.
O diagnóstico é baseado em achados clínicos. A biópsia de um dos nódulos pode confirmar a suspeita clínica, mas não se pode basear-se apenas na anatomia patológica, uma vez que os neuromas têm histologia comum6.
O tratamento limita-se a excisão dos tumores que causam sintomas, como dor, ou incapacidade funcional, ou que estejam levando a alterações estéticas e psicológicas ao paciente. Também se indica ressecção de lesões com crescimento acelerado com suspeita de malignidade6, visto que a transformação sarcomatosa está presente em 5 a 10% dos casos.
CONCLUSÃO
O atendimento dos pacientes com NF1 mostra que a doença pode apresentar grande impacto na qualidade de vida do paciente, tanto do ponto de vista clínico quanto no aspecto psicológico.
As ressecções completas são a melhor opção de tratamento, mas podem ser difíceis em pacientes com grandes massas tumorais, como no presente caso, pela falta de um plano de clivagem entre o tumor e os tecidos vizinhos. Pode ser um desafio ao cirurgião plástico, que deve estar atento as manifestações variadas desta afecção. São aceitáveis ressecções parciais, quando a total não puder ser realizada.
REFERÊNCIAS
1. Huson SM, Harper PS, Compston DA. Von Recklinghausen neurofibromatosis. A clinical and population study in south-east Wales. Brain. 1988;111 (Pt 6):1355-81. DOI: http://dx.doi.org/10.1093/brain/111.6.1355
2. Friedman JM. Epidemiology of neurofibromatosis type 1. Am J Med Genet. 1999;89(1):1-6. PMID: 10469430 DOI: http://dx.doi.org/10.1002/(SICI)1096-8628(19990326)89:1<1::AID-AJMG3>3.0.CO;2-8
3. Gottfried ON, Viskochil DH, Fults DW, Couldwell WT. Molecular, genetic, and cellular pathogenesis of neurofibromas and surgical implications. Neurosurgery. 2006;58(1):1-16. DOI: http://dx.doi.org/10.1227/01.NEU.0000190651.45384.8B
4. MacCollin M. Neurofibromatosis 2 — Clinical aspects. In: Friedman JM, Gutmann DH, Collin M, Riccardi VM. Phenotype, Natural History, and Pathogenesis. 3rd ed. Baltimore: The Johns Hopkins University Press; 1999. p. 299-326.
5. Baser ME, Friedman JM, Evans DG. Increasing the specificity of diagnostic criteria for Schwannomatosis. Neurology. 2006;66(5):730-2. DOI: http://dx.doi.org/10.1212/01.wnl.0000201190.89751.41
6. Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44(2):81-8. PMID: 17105749 DOI: http://dx.doi.org/10.1136/jmg.2006.045906
7. North KN. Clinical aspects of neurofibromatosis 1. Eur J Paediatr Neurol. 1998;2(5):223-31. DOI: http://dx.doi.org/10.1016/S1090-3798(98)80035-4
8. Ruggieri M, Huson SM. The neurofibromatoses. An overview. Ital J Neurol Sci. 1999;20(2):89-108. PMID: 10933430 DOI: http://dx.doi.org/10.1007/s100720050017
9. Fit Z, Patrick BT, Ejsen AZ, Wolff K, Freedberg JM, Austen KF. Dermatology in general medicine. New York: McGraw Hill; 1979.
Hospital Universitário, Universidade Federal de Santa Catarina, Florianópolis, SC, Brasil
Endereço Autor:
Inara do Carmo Lucchese
Rua Professora Maria Flora Pausewang, s/n, Trindade
Florianópolis, SC, Brasil - CEP 88036-800
E-mail: iclucchese@gmail.com
 All scientific articles published at www.rbcp.org.br are licensed under a Creative Commons license
All scientific articles published at www.rbcp.org.br are licensed under a Creative Commons license


