


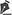
Special Article - Year 2006 - Volume 21 -
Burn victim with Behçet´s disease in association with trisomy 8: case report and literature review
Queimadura em Paciente com Doença de Behçet Associada a Trissomia do Cromossomo 8: Relato De Caso E Revisão De Literatura
ABSTRACT
Behçet's syndrome is a rare systemic vasculitis which etiology is still unknown. It is characterized by recurrent episodes of orogenital ulcers, systemic vasculitis, systemic and retinal venous thrombosis. About 10 cases of Behçet's syndrome associated with myelodisplasic syndrome were described in medical literature. This paper describes a burned patient with Behçet's syndrome associated with chromosome 8 trisomy and myelodisplasic syndrome, with fatal evolution. A literature revision about Behçet's disease is also reported.
Keywords: Burns. Behcet syndrome. Trisomy. Chromosomes, human, pair 8
RESUMO
A síndrome de Behçet é uma rara vasculite sistêmica, de etiologia desconhecida, que se apresenta com episódios recorrentes de aftas orogenitais, vasculite sistêmica, tromboses venosas retinianas e sistêmicas. Foram descritos na literatura cerca de 10 casos associados com síndrome mielodisplásica. Neste trabalho, relata-se o caso clínico de um paciente com diagnóstico de doença de Behçet, associada à trissomia do cromossomo 8 e síndrome mielodisplásica, que teve como agravante uma queimadura por escaldadura, com evolução fatal. Também foi realizada uma revisão de literatura sobre a doença de Behçet.
Palavras-chave: Queimaduras. Síndrome de Behçet. Trissomia. Cromossomos humanos par 8
A síndrome de Behçet é uma vasculite sistêmica, de etiologia desconhecida, que afeta caracteristicamente vênulas1. É caracterizada por episódios recorrentes de aftas orogenitais, vasculite sistêmica, tromboses venosas retinianas e sistêmicas2. Hulusi Behçet, professor de Dermatologia em Istambul, descreveu a tríade de úlceras aftosas na boca e genitália, uveíte recorrente e lesões dermatológicas3. Outras manifestações incluem as neurológicas, cardiovasculares, gastrointestinais e musculoesqueléticas.
O atual critério diagnóstico determina a associação de úlceras orais recorrentes (3 ou mais vezes ao ano) a dois dos seguintes sintomas, na ausência de outra doença sistêmica: úlceras genitais recorrentes, lesões oculares (uveíte ou vasculite retiniana), lesões de pele (eritema nodoso, pseudofoliculite, lesões papulo-pustulares, nódulos acneiformes) ou teste positivo para patergia4. A doença de Behçet pode ser classificada em completa ou incompleta. Na maioria das vezes, não envolve lesões oculares e é considerada incompleta.
Sua etiologia permanece obscura; infecções virais5 e bacterianas foram aventadas como alguns dos fatores etiológicos.
A associação com a síndrome mielodisplásica é menos freqüente. Cerca de dez casos já foram relatados, a maioria em pacientes japoneses6.
Este artigo descreve um paciente com diagnóstico de síndrome de Behçet associada a trissomia do cromossomo 8, hipertensão arterial sistêmica, insuficiência cardíaca congestiva e diabetes mellitus, que teve suas doenças de base agravadas por uma queimadura.
RELATO DO CASO
J.S., 71 anos, sexo masculino, pardo, natural e procedente de São Paulo. Portador de hipertensão arterial sistêmica tratada com vasodilatador, diabetes mellitus tipo II, em uso de hipoglicemiantes orais, e insuficiência cardíaca congestiva, em seguimento ambulatorial na Unidade Básica de Saúde. Apresentava também como antecedentes angiodisplasia total dos cólons (diagnosticada em outro serviço após vários episódios de hemorragia digestiva baixa), além de um prévio episódio de acidente vascular cerebral.
Encaminhado para o Serviço de Hematologia do Hospital das Clínicas da FMUSP, recebeu o diagnóstico de Doença de Behçet, decorrente de achados como hipoagregação plaquetária, mielograma compatível com síndrome mielodisplásica e cariótipo com trissomia do cromossomo oito.
Em 13 de janeiro de 2002, sofreu queimadura por água fervente. Após o primeiro atendimento (em hospital primário) e, somente no 3º dia pós-queimadura, foi transferido para serviço especializado no Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo. Ao exame físico encontrava-se descorado, com edema em membros inferiores, queimaduras de primeiro (1% em pescoço, 0,5% em antebraço e 2% em pernas) e segundo graus (6% em coxas), com exsudato purulento, totalizando 9,5% de área queimada (Figura 1). Radiografia de tórax mostrava área cardíaca aumentada. Exames laboratoriais revelavam anemia (Hb = 10,5), leucocitose (30 mil) e coagulopatia (TP = 45,5%).
Figura 1 - Paciente apresentava 9,5% de superfície corpórea queimada, segundo mostrado pelo gráfico de Lund and Browder.
Após analgesia, limpeza e desbridamento, o paciente foi encaminhado à UTI do Serviço de Queimaduras do HCFMUSP por seus antecedentes patológicos, onde se iniciou o tratamento local da queimadura e antibioticoterapia sistêmica com oxacilina e ciprofloxacina.
Ecocardiograma revelou comprometimento difuso do miocárdio e discreto derrame pericárdico; Doppler com discreto fluxo retrógrado e turbulento em valvas mitral e aórtica.
No 14º dia de internação, apresentou melena. Exames laboratoriais revelaram piora da anemia (Hb = 7,9; Ht = 23,4%), plaquetopenia (35 mil) e TP = 50,7%. Foi transfundido com concentrado de hemácias e submetido a enxertia de pele parcial em ferida da coxa, que havia apresentado aprofundamento da queimadura. Iniciou-se, também, administração de plasma congelado e prednisona via oral.
No 5º pós-operatório, o enxerto estava com 100% de pega e o paciente saiu da UTI (Figura 2).
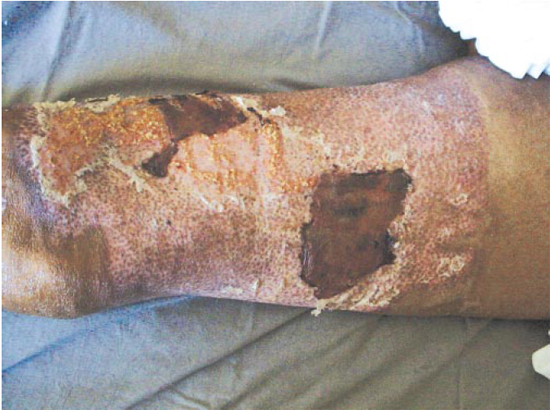
Figura 2 - Quinto dia pós-enxertia de pele de espessura parcial, em ferida da coxa.
Por volta do 20º dia de internação, apresentou convulsão tônico-clônica generalizada, tratada inicialmente com benzodiazepínicos. Tomografia de crânio revelou duas coleções subdurais, sendo a maior à esquerda, compatível com hematoma subdural crônico, com sinal de trepanação prévia e efeito de massa. Foi introduzida fenitoína e o paciente mantido em observação, mas como apresentou rebaixamento do nível de consciência, foi submetido à trepanação parietal esquerda e drenagem do hematoma subdural, sem intercorrências.
Evoluiu desfavoravelmente, com piora da função renal, isolamento de vários microorganismos em culturas, persistência da hemorragia digestiva baixa (cuja endoscopia não revelou sinais de sangramento) e CIVD e, finalmente, com insuficiência respiratória e parada cardiorrespiratória não responsiva às manobras de ressuscitação, em 3 de março de 2002.
Durante toda a internação, o paciente foi transfundido com um total de 173 unidades de plasma fresco congelado, 40 unidades de concentrado de hemácias e três unidades de plaquetas.
DISCUSSÃO
A síndrome de Behçet é caracterizada por estomatite aftosa recorrente, uveíte, úlceras genitais e lesões dermatológicas7 e leva o nome do autor que a primeiro descreveu, em 1937. Esta entidade, originalmente restrita à tríade clínica descrita acima, é considerada, atualmente, uma doença inflamatória sistêmica, que pode afetar vários órgãos, incluindo mucosa (estomatite aftosa), genitais (úlceras), olhos (uveíte e vasculite da retina), vasos (trombose de veias profundas, constrição arterial - vásculo-Behçet), sistema nervoso (neuro-Behçet) e intestino (úlceras "punched-out" - Behçet intestinal). As manifestações gastrointestinais são comuns e, em algumas revisões, apresentadas como distúrbios inespecíficos de desconforto abdominal, distensão, náusea e diarréia. A maioria dos pacientes com sintomas intestinais da doença de Behçet não possui lesões oculares, sendo a mesma classificada como incompleta.
Como são comuns as manifestações vasculares, classificase como vasculite. A histopatologia predominante é de infiltração de linfócitos e monócitos e, às vezes, leucócitos polimorfonucleares, por meio de vênulas sem alterações microscópicas em sua parede8. A vasculite, principal processo patológico da doença, pode ser parcialmente explicada pelo fenômeno trombótico9. A trombose pode resultar de uma combinação de anormalidades de procoagulantes, anticoagulantes e fatores fibrinolíticos, juntamente com a vasculite e lesão endotelial10. A doença de Behçet tem prevalência aumentada em países ao longo da antiga Rota da Seda, do Japão à região Mediterrânea. A prevalência varia de 1:10.000, no Japão, até 1:500.000, na América do Norte e Europa.
Seu início é geralmente na terceira década de vida, mas pode ocorrer em qualquer idade. A relação homem:mulher é equivalente (1:1), no Japão e na Europa, mas aumenta significantemente (1,5-5:1), em alguns países Mediterrâneos e no Oriente Médio11. A patogenia da doença ainda não é bem estabelecida, mas a hipótese mais aceita é a de uma resposta inflamatória intensa, deflagrada por algum agente infeccioso em um hospedeiro susceptível. Alguns estudos demonstram maior prevalência de um tipo incomum de Streptococcus sanguis na flora oral desses pacientes12.
Anticorpos séricos contra HSV1 foram encontrados em maior proporção em pacientes com Behçet do que em controles saudáveis, e vários estudos mostraram evidências de que o HLA-B51 está fortemente associado a essa doença em diferentes grupos étnicos13.
A síndrome de Behçet abrange amplo espectro de manifestações clínicas e é caracterizada por exacerbações imprevisíveis e períodos de remissão. As úlceras orais costumam ser os sintomas iniciais na maioria dos casos e podem ocorrer em 97 a 100% dos pacientes. As úlceras genitais são menos freqüentes, ocorrendo em 60-80% dos casos14. As lesões cutâneas ocorrem em aproximadamente 80% dos pacientes e são divididas em eritema nodoso e lesões papulo-pustulares/acneiformes15. O envolvimento ocular é bilateral, na maioria dos casos, podendo ser assimétrico.
Neuro-Behçet acomete cerca de 5% dos casos. A clínica inclui sintomas piramidais bilaterais, alterações do nível de consciência, hemiparesia, paralisia de nervos cranianos e distúrbios esfincterianos na maioria dos pacientes16. No caso em questão, as alterações do nível de consciência pareceram decorrer do hematoma subdural crônico e, posteriormente, do delirium, apesar de não ter sido realizada ressonância magnética, normalmente mais sensível do que a tomografia computadorizada, que poderia revelar lesões pseudotumorais, lesões nos gânglios da base, múltiplas lesões na substância branca ou na medula espinhal, comuns na neuro-Behçet.
O envolvimento gastrointestinal varia de acordo com as populações estudadas, sendo comum nos pacientes japoneses. As úlceras ocorrem predominantemente na região ileocecal13, mas podem ser vistas em todo o trato intestinal, além do esôfago, causando disfagia, dor abdominal e diarréia (podendo ser sanguinolenta), até perfuração intestinal e fístulas perianais. Além disso, a recorrência da doença em áreas de anastomoses cirúrgicas é comum17. O paciente em questão apresentava histórico de repetidos episódios de enterorragia, inicialmente creditados à angiodisplasia dos cólons diagnosticada por colonoscopia prévia à internação.
Apesar de apresentar-se como vasculite sistêmica, os rins costumam ser poupados. Discreta proteinúria e hematúria microscópica podem ocorrer em alguns pacientes. A causa mais comum de falência renal na doença de Behçet é a amiloidose18.
O envolvimento cardíaco é incomum, mas casos de pericardite, derrame pericárdico, acometimento valvar, trombose coronária e aneurismas19 já foram documentados. O paciente em questão apresentava derrame pericárdico e sinais de comprometimento valvar, segundo ecocardiograma; tais achados, provavelmente, fariam parte do quadro de insuficiência cardíaca congestiva prévia, não se podendo afirmar relação causal entre esta e a doença de Behçet.
Esta doença, associada à síndrome mielodisplásica, já foi relatada em dez casos, que mostraram, em sua maioria, associação também com a trissomia do cromossomo 8. Anormalidades cromossômicas em pacientes com síndrome mielodisplásica ocorrem em 37 a 51% dos casos, nos quais a trissomia do 8 é responsável por 10 a 30%. Ohno et al.6 sugeriram que a trissomia do cromossomo 8 poderia predispor à doença de Behçet em um subgrupo das síndromes mielodisplásicas, já que a porcentagem de trissomia do 8 em pacientes desse subgrupo com doença de Behçet era maior do que nos pacientes portadores de síndrome mielodisplásica isolada.
O tratamento da doença de Behçet é problemático e inclui o uso de diversas drogas, geralmente em associação, que costuma ser mais eficaz do que com o uso de uma só droga20. Algumas das drogas usadas incluem ciclosporina, talidomida, interferon, aciclovir, corticosteróides, ciclofosfamida, tacrolimus, metotrexate, azatioprina e colchicina.
O curso clínico da doença é variado, dificultando o prognóstico em longo prazo. Fatores como sexo masculino e início precoce dos sintomas contribuem separadamente para um curso clínico mais grave. É sabido também que homens com envolvimento ocular apresentam pior prognóstico. Envolvimento do sistema nervoso central, artérias ou veias, e perfurações gastrintestinais parecem estar também associadas a pior prognóstico.
No caso apresentado, a queimadura descompensou as doenças de base do paciente; a infecção do local queimado, por sua vez, ajudou a deflagrar a descompensação da síndrome mielodisplásica, resultando em melena e sangramento no sistema nervoso central. Na evolução, o paciente apresentou piora, tanto da anemia quanto da plaquetopenia, e as transfusões recebidas não foram suficientes para evitar a falência de seu estado clínico, culminando com o óbito.
CONCLUSÃO
A síndrome de Behçet representa uma doença sistêmica crônica de múltiplas manifestações, difícil diagnóstico e, principalmente, complexo manejo clínico. O paciente em questão apresentava a rara associação entre doença de Behçet e síndrome mielodisplásica (apenas 12 casos relatados na literatura), além de inúmeros fatores de gravidade (idade avançada, insuficiência cardíaca congestiva e diabetes mellitus), sendo a queimadura um fator agravante da delicada situação clínica em que se encontrava.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Kontogiannis V, Powell RJ. Behçet´s disease. Postgrad Med J. 2000;76(900):629-37.
2. Verity DH, Wallace GR, Vaughan RW, Stanford MR. Behçet´s disease: from Hippocrates to the third millennium. Br J Ophthalmol. 2003;87(9):1175-83.
3. James DG. 'Silk route disease' (Behçet´s disease). West J Med. 1998;148(4):433-7.
4. Al-Otaibi LM, Porter SR, Poate TW. Behçet´s disease: a review. J Dent Res. 2005;84(3):209-22.
5. Sohn S. Etiopathology of Behçet´s disease: herpes simplex virus infection and animal model. Yonsei Med J. 1997;38(6):359-64.
6. Ohno E, Ohtsuka E, Watanabe K, Kohno T, Takeoka K, Saburi Y, et al. Behçet's disease associated with myelodysplastic syndromes. A case report and a review of the literature. Cancer 1997;79(2):262-8.
7. Hirohata S, Kikuchi H. Behçet´s disease. Arthritis Res Ther. 2003;5(3):139-46.
8. Sakane T, Suzuki N, Nagafuchi H. Etiopathology of Behçet´s disease: immunological aspects. Yonsei Med J. 1997;38(6):350-8.
9. Sakane T, Takeno M, Suzuki N, Inaba G. Behçet's disease. N Engl J Med. 1999;341(17):1284-90.
10. Leiba M, Sidi Y, Gur H, Leiba A, Ehrenfeld M. Behçet´s disease and thrombophilia. Ann Rheum Dis. 2001; 60(12):1081-5.
11. Atmaca LS, Idil A, Batioglu F. A descriptive study in Behçet´s disease. Acta Ophthalmol Scand. 1996;74(4):403-6.
12. Yoshikawa K, Kotake S, Matsuda H. Behçet's disease and streptococcal antigens. Nippon Ganka Gakkai Zasshi. 1996;100(3):173-80.
13. Jankowski J, Crombie I, Jankowski R. Behçet's syndrome in Scotland. Postgrad Med J. 1992;68 (801):566-70.
14. Yazici H, Yurdakul S, Hamuryudan V. Behçet's syndrome. Curr Opin Rheumatol. 1999;11(1):53-7.
15. Lee ES, Bang D, Lee S. Dermatologic manifestation of Behçet´s diasease. Yonsei Med J. 1997;38(6):380-9.
16. Serdaroglu P. Behçet's disease and the nervous system. J Neurol. 1998;245(4):197-205.
17. Chung SY, Ha HK, Kim JH, Kim KW, Cho N, Cho KS, et al. Radiologic findings of Behçet syndrome involving the gastrointestinal tract. Radiographics. 2001;21(4):911-26.
18. Akpolat T, Diri B, Oguz Y, Yilmaz E, Yavuz M, Dilek M. Behçet´s disease and renal failure. Nephrol Dial Transplant. 2003;18(5):888-91.
19. Ioakimidis D, Georganas C, Panagoulis C, Gournizakis A, Iliopoulos A, Kremastinos D, et al. A case of Adamantiades- Behçet's syndrome presenting as myocardial infarction. Clin Exp Rheumatol. 1993;11(2):183-6.
20. Bang D. Treatment of Behçet´s disease. Yonsei Med J. 1997;38(6):401-10.
I. Médica Colaboradora da Divisão de Cirurgia Plástica e Queimaduras do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo.
II. Médico Residente da Divisão de Cirurgia Plástica e Queimaduras do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo.
III. Médico Assistente da Divisão de Cirurgia Plástica e Queimaduras do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo.
IV. Assistente Doutor; médico responsável pelo Serviço de Queimaduras da Divisão de Cirurgia Plástica e Queimaduras do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo.
V. Professor Titular da Disciplina de Cirurgia Plástica da Faculdade de Medicina da Universidade de São Paulo.
Correspondência para:
Julia Peres
Al. dos Jauaperis, 1.083 - apto. 66
CEP: 04523-014
Tel: 0xx11 8596-9673
E-mail: peresjulia@hotmail.com
Trabalho realizado no Serviço de Queimaduras do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, SP.
Artigo recebido: 20/11/2006
Artigo aprovado: 14/12/2006
 Read in Portuguese
Read in Portuguese
 Read in English
Read in English
 PDF PT
PDF PT
 Print
Print
 Send this article by email
Send this article by email
 How to Cite
How to Cite
 Mendeley
Mendeley
 Pocket
Pocket