


Case Report - Year 2015 - Volume 30 -
Proteus Syndrome: case reports
Síndrome de Proteus: relato de casos
ABSTRACT
INTRODUCTION: Proteus syndrome is a complex and rare disorder classified as a hamartomatous disease. It was first described in two patients in 1979, by Cohen and Hayden. Proteus syndrome is difficult to diagnose, and is often confused with Klippel-Trenaunay-Weber syndrome, neurofibromatosis, or Sturge-Weber syndrome. In this study we describe two patients who were treated at the Plastic and Reconstructive Surgery Service of the Federal University of Paraná.
METHOD: A 6-year-old male patient (case 1) presented to the Service with lipomatosis and asymmetry, as the primary findings. A 20-year-old (case 2) was admitted to the Service with a diagnosis of Klippel-Trenaunay-Weber syndrome, which later was shown to be Proteus syndrome.
CONCLUSION: The etiological hypothesis that is most accepted for this disease is genetic. It is believed that somatic mosaicism may occur during pathogenesis, which can be lethal in the mosaic state. Premature death is common. However, the most common sequelae are the occurrence of unusual tumors. The care of patients with this syndrome is a challenge due to medical and psychosocial consequences.
Keywords: Proteus syndrome; Hamartoma; Lipoma.
RESUMO
INTRODUÇÃO: A síndrome de Proteus é uma doença complexa e rara, classificada nos grupo das hamartoses. Foi primeiramente descrita em dois pacientes, em 1979, por Cohen e Hayden. Existe dificuldade no diagnóstico, sendo comum a confusão com síndromes de Klippel-Trenaunay-Weber, neurofibromatose ou Stuge-Weber. Apresentamos dois casos tratados no Serviço de Cirurgia Plástica e Reparadora da Universidade Federal do Paraná.
MÉTODO: Paciente masculino (caso 1), que chegou ao serviço aos 6 anos de idade, tendo como principais apresentações lipomatoses e assimetrias. A segunda paciente (caso 2) deu entrada no serviço com 20 anos de idade e diagnóstico de síndrome de Klippel-Trenaunay-Weber, que posteriormente mostrou se tratar de síndrome de Proteus.
CONCLUSÃO: A hipótese etiológica mais aceita para a doença é genética. Acredita-se que exista mosaicismo somático e que a doença seja letal no estado não mosaico. Morte prematura é bastante frequente. Entretanto, a sequela mais comum é a ocorrência de tumores incomuns. O cuidado dos pacientes portadores da síndrome é um desafio devido às suas consequências médicas e psicossociais.
Palavras-chave: Síndrome de Proteus; Hamartoma; Lipoma.
Proteus syndrome is a complex and rare disorder classified as a hamartomatous disease. It was first described in two patients in 1979 by Cohen and Hayden1. In 1983, Wiedemann et al.2 named this disease Proteus syndrome, after the Greek god who could change his body shape; this is reflected in the variability of the clinical manifestations of this syndrome. The etiology of Proteus syndrome is still not fully clarified. However, it is believed that genetic causes may be involved, due to the multisystemic nature of the pathology3. Proteus syndrome is difficult to diagnose, due to its low incidence and manifestations similar to other disorders, such as Klippel-Trenaunay-Weber syndrome, neurofibromatosis, encephalocraniocutaneous lipomatosis (ECCL), Maffucci syndrome, Sturge-Weber syndrome, and Bannayan-Zonana syndrome4-7.
The manifestations of Proteus syndrome may vary, with the occurrence of a series of tumors of disproportionate size and asymmetrical growth, developing in different parts of the body; in particular, these result in bone tumors, cerebriform connective tissue nevi, epidermoid nevi, vascular malformations, and irregular deposits of adipose tissue. A number of other manifestations may also occur, including splenomegaly, digital hypertrophy, scoliosis, lip and finger hypertrophy, heart disease, cystic fibrosis, and mental retardation2,3,8,9. The clinical symptoms may appear at birth, but are more prominent during the postnatal development of these patients9. Few investigations address the therapeutic modalities that can benefit these patients. In this study, we present three patients with Proteus syndrome, and discuss their diagnosis and treatment.
CASE REPORT
Case 1
A 6-year-old male patient was referred to the Plastic Surgery Service of the Hospital de Clínicas. He presented with disseminated lymphangiomas on his back and lower limbs that started to develop at the age of 5 months, with subsequent progressive growth in volume. The patient had healthy non-consanguineous parents. Both parents were 31 years old at the time of conception, and this was the mother's third gestation. His older sister died two hours after birth, and his younger brother died 3 days after birth. There were no other similar cases in the family. Cesarean delivery was performed at the 32nd week of pregnancy, but no reports indicated the necessity for early delivery. At birth, he weighed 2,730 grams, and was hospitalized for 7 days due to neonatal jaundice. At 4 months of age, he was circumcised, and an inguinal hernia was repaired. He had delayed psychomotor development, sat without support at 8 months, and only walked without assistance at 2 years.
At 6 years of age, the following characteristics were observed: weight 36.5 kg, height 128.5 cm, head circumference 55 cm, ear length 6.5 cm, interpupillary distance 5.5 cm, intermammary distance 15.5 cm, right leg 25 cm, and left leg 28.5 cm. We observed large dorsal tumors with a soft consistency, greater body asymmetry on the left side, pes planus and hallux varus, and a left lower limb shortened by 1 cm, with circumference increased by 1 cm. The electrocardiogram showed sinus arrhythmia. There were no cranial abnormalities or signs of mental retardation. Genetic evaluation showed a 46 XY karyotype. Accordingly, he was diagnosed with Proteus syndrome (Figures 1A and 1B).
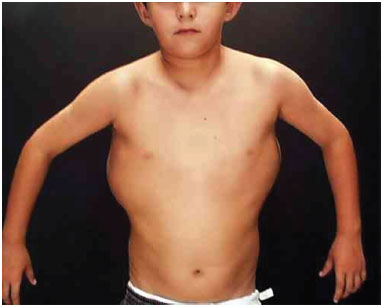
Figure 1A. The patient presents disseminated lymphangiomas on the back.
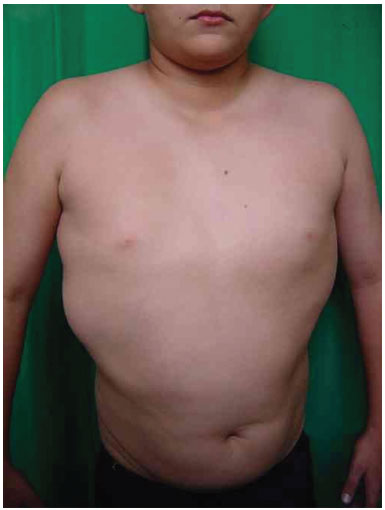
Figure 1B. The patient presents disseminated lymphangiomas on the back.
Magnetic resonance imaging (MRI) revealed the presence of tumors above and below the latissimus dorsi muscle; the diameter of blood vessels supplying the tumors was around 0.5 cm. We chose to perform ultrasonic liposuction, due to a report in the literature of a lower probability of bleeding. The procedure was performed in April 2001, with the removal of 600 mL from each side, partial resection of the tumor tissue, and a satisfactory postoperative outcome. However, large tumors were still present on the back, and the result was considered inadequate. In 2003, we carried out a second uncomplicated procedure, with resection of a 1,434 g mass, which included the muscles of the right back. Pathological results confirmed lipomatosis (Figures 1C and 1D). In June 2003, the patient underwent resection of a tumor weighing 1,350 g in the left dorsal region. This had the same pathological characteristics. In January 2006, he underwent additional fatty tumor resection between muscle planes, and below the latissimus dorsi muscle. The resected fragments weighed 175 g and presented the same pathological features. Postoperative complications were characterized by local inflammatory signs. However, a favorable outcome was achieved with the use of cephalexin and analgesics.
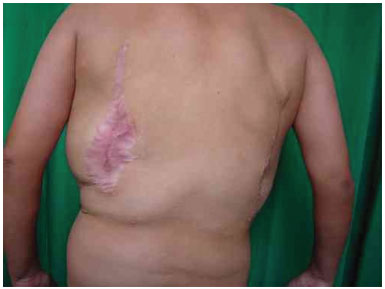
Figure 1C. Postoperative appearance of tumor resection.
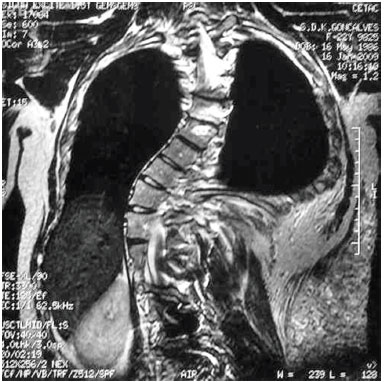
Figure 1D. Magnetic resonance imaging demonstrating the presence of tumors above and below the latissimus dorsi muscle.
Case 2
A 20-year-old female patient was referred to the Plastic Surgery Service of the Hospital de Clínicas of Curitiba, due to the development of lipomas on her back, which developed at 6 months of age. She had healthy, non-consanguineous parents. The mother was 21 years old and the father 22 at the time of conception. Pregnancy was uncomplicated. A cesarean delivery was performed at term. Birth weight was 3,100 g, length 45 cm, and head circumference 34 cm. She developed neonatal jaundice, which resolved with phototherapy. She had two normal brothers. There are no reports of illness or other similar cases in the family. We observed three tumors with soft consistency on the left dorsal region, which were mobile, painless, and non-adherent, and with a diameter ranging from 8 to 10 cm. Hypertrophy of the right lower limb was also observed, with a difference of 2 cm in the circumference of the right vs. the left thigh, and of the right vs. the left leg. She presented with a capillary vascular malformation on the left lateral trunk.
She was first treated by the orthopedic service for a complaint of asymmetry regarding the right lower limb, and thoracolumbar kyphoscoliosis, which was treated with arthrodesis and Harrington rods. She was evaluated in the pediatric surgery service for the lipomas. The initial diagnosis was Klippel-Trenaunay-Weber syndrome, and the initial radiographs showed no bone abnormalities.
In this syndrome, the presence of a port-wine stain, varicose veins, and bone hypertrophy occur in 98-100% of cases, and the port-wine stain and vascular changes are usually present in the hypertrophied body part. Klippel-Trenaunay-Weber syndrome is a congenital malformation that can arise due to an autosomal dominant mutation during embryogenesis, or autosomal dominance with incomplete penetrance. This explains familial occurrence, but does not present a pattern of regular generational frequency, as the syndrome can be transmitted for many generations by phenotypically normal individuals10.
The genetic evaluation was based on history. Clinical evaluation showed dorsal lipomatosis, a capillary malformation, scoliosis, and asymmetric feet (the right foot was larger). As there are no relevant laboratory tests, the diagnosis of Proteus syndrome was based on clinical and family data. The electrocardiogram showed sinus arrhythmia.
Three surgical procedures were carried out to remove the dorsal tumors. The first procedure was performed when the patient was 11 years old, and used liposuction to remove lipomas. When evaluated by the plastic surgery service, a generalized hypertrophy of the fatty tissue was observed. Twelve years after the first surgery, the patient presented with recurrence of lipomatous lesions. Computed tomography showed several cystic images in the left hemithorax, which appeared tubular, with internal vascular flow, and were compatible with a venous malformation. A second procedure for removal of a mass from the posterior region was performed, with resection of a 200 g tumor with lobulated appearance, measuring 17 cm in its greatest length; pathological examination was compatible with a giant lipoma. The patient ultimately underwent back liposuction. In 2006, the patient was seen as an outpatient, with stability of the tumors and vascular malformation (Figure 2 A and 2 B).
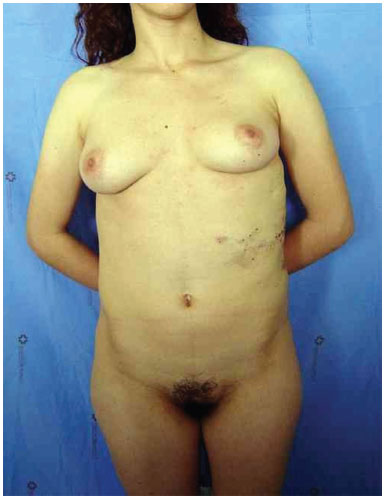
Figure 2A. Patient with lipomatosis on the chest.
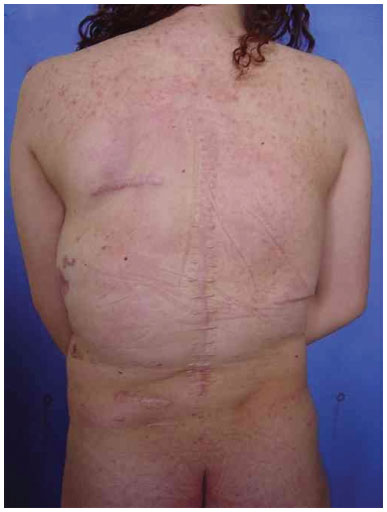
Figure 2B. Postoperative appearance.
Case 3
A 19-month-old male patient was referred to the Plastic Surgery Service of the Hospital de Clínicas, due to a massive lymphangioma in the flank region and right hypochondrium, extending to the dorsal region. The parents were healthy and non-consanguineous, and this was the mother's first gestation. There were no other similar cases in the family (Figure 3 A, B, C, and D).
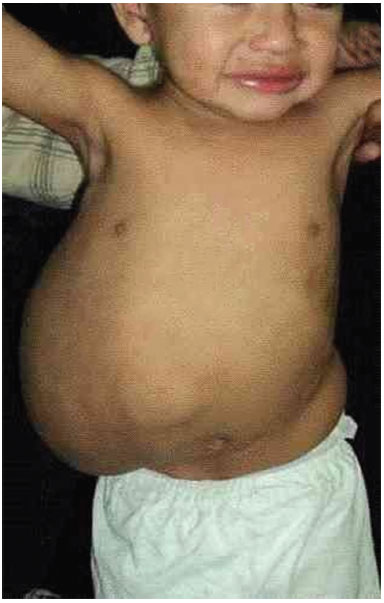
Figure 3A. Patient with large lymphangioma in the flank and right hypochondrium extending to the dorsal region.
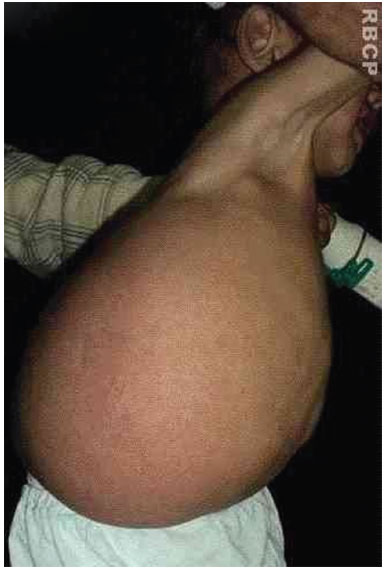
Figure 3 B. Large lymphangioma in the flank and right hypochondrium extending to the dorsal region.
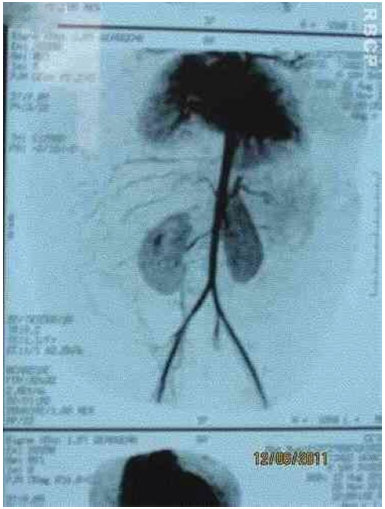
Figure 3C. Arteriographic appearance of the lesion.
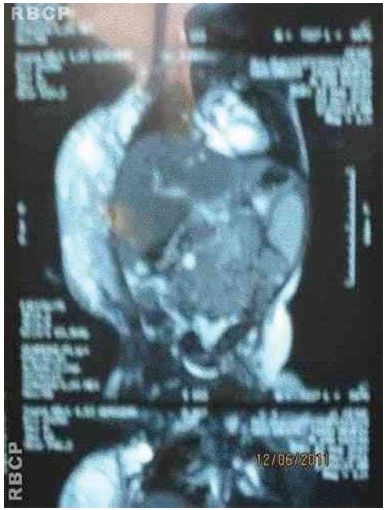
Figure 3D. MRI of the tumor.
The patient was admitted as an emergency with septic shock, but no definite focus. He was treated with a broad-spectrum antibiotic. With the assumption that the focus was a lymphangioma, we decided to wait for an improvement in the clinical condition before planning surgery.
A complete tumor resection was performed, with dissection down to the rectus abdominis muscle, but without entry into the abdominal cavity; the fifth rib marked the upper limit, the scapula the posterior limit, and the inguinal region the lower limit of the tumor. A full-thickness skin graft was removed from the resected tumor, and grafted during the same surgery (Figure 3 E, F, G and I).
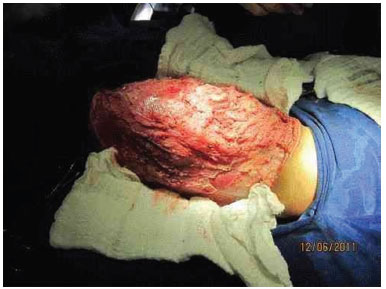
Figure 3E. Intraoperative appearance, during removal of the tumor.

Figure 3F. Preparation for the removal of the skin graft to cover the defect caused by tumor excision.
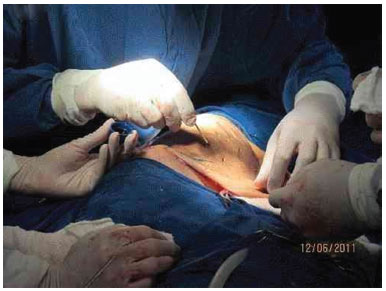
Figure 3G. Fixation appearance of the skin graft.
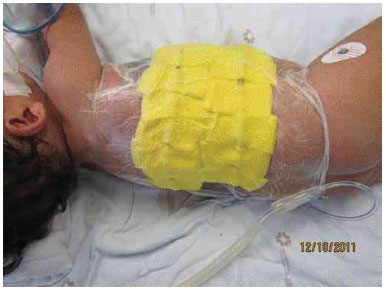
Figure 3H. Positioning of the vacuum dressing, after graft placement.
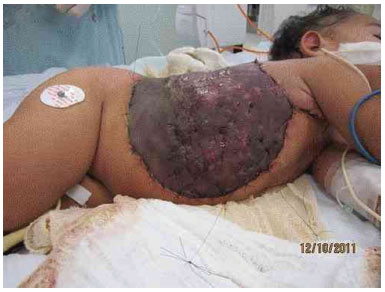
Figure 3I. Graft appearance 7 days after surgery.
To improve graft integration, and due to the possible local drainage of large volumes of lymphatic fluid, we placed a vacuum dressing on the graft (Figure 3 H).
The patient was sedated and intubated in the intensive care unit for 7 days, in order to maintain proper position and prevent graft movement. The patient presented satisfactory graft integration and favorable clinical progress, and was subsequently discharged from the hospital.
DISCUSSION
Proteus syndrome is characterized by a polymorphic phenotype. The clinical findings change over time, making this syndrome difficult to diagnose. Proteus syndrome is a rare disease, and its natural history is still not fully understood5. The etiological hypothesis that is most accepted for this disease is genetic, and is believed to represent the existence of somatic mosaicism. The disease is lethal in a non-mosaic state. The cases are mainly sporadic. Three adults have been reported in the literature to be affected by the disease, and subsequently gave birth to normal children9. In several cases, asymmetry and hemihypertrophy were observed at birth, although these are much more prominent during postnatal development. The overgrowth of bone and soft tissues tends to reach a plateau after adolescence. The final height of the patients, as well as their growth during adolescence, seems to be normal8,11-13.
The main sequelae of this syndrome are premature death and the occurrence of unusual tumors. Premature death is mainly due to deep venous thrombosis, resulting in pulmonary embolism. Attention is drawn to the lipomas, which are histologically benign tumors, but which present with invasive intra-abdominal or intrathoracic behavior8.
Due to the difficulty in identifying this syndrome, diagnostic criteria were established at the first National Conference on Proteus Syndrome in 1998. To make a diagnosis, all these criteria should be present: mosaic distribution of the lesions, progressive course, and sporadic occurrence of the disease (i.e., not familial)8.
Different manifestations are possible during development of this syndrome. Among the most common are hemihypertrophy, skull hyperostosis, cerebriform nevi, pigmented nevi, subcutaneous tumors, vascular malformations, abnormal adipose tissue, central nervous system manifestations, and varied ophthalmological symptoms. Other less common manifestations include craniofacial abnormalities, facial phenotypes, tumors, splenomegaly, and thymic hypertrophy8,9.
Hemihypertrophy usually develops during childhood, and evolves through late adolescence. It can be partial, complete, or mixed, leading to limb dysfunction and difficulty walking. Treatment includes epiphysiodesis, arthrodesis, limb shortening or lengthening, and asymmetry reduction. Cerebriform nevi are lesions composed of connective tissue with excess collagen. These most commonly occur on the plantar surface and palm. These are not mandatory for diagnosis, but when present are almost pathognomonic of the syndrome.
Subcutaneous tumors (lipomas, hemangiomas, and lymphangiomas) develop differently and in any part of the body. They can grow to the point of infiltrating local tissues, and cause a difficult resection. The treatment includes resection, dissection, and liposuction. The results are often unsatisfactory, due to the recurrence and formation of hypertrophic scars6,11. Lipomas are composed primarily of mature adipocytes. Although the histopathologic appearance of these tumors is benign, they may present with aggressive behavior8, depending on their location, especially if intrathoracic or intra-abdominal. An increase or decrease of fat deposits can be found in the same patient, and in different parts of the body, demonstrating impaired regulation of adipose tissue. A different pattern seems to occur with lipomas and increases in subcutaneous fat. Adipose tissue deposits can be found between the muscles of these patients14. This was apparent in two cases. In case 1, we observed infiltration of the large dorsal muscles.
Asymmetrical growth is seen, especially in the lower limbs, and one study compared thighs by using MRI. We often noted an excessive increase in the subcutaneous fat of the enlarged limb14. Vascular malformations may have only one component (e.g., capillary, lymphatic, or venous), or may be combined (e.g., capillary and venous, or capillary, venous and lymphatic). They develop in proportion with the patient, never involute, and can expand8. The less common tumors include ovarian cystadenomas, meningiomas, testicular tumors, and parotid adenomas9.
The management of these patients should be performed by a multidisciplinary team. Examinations should be conducted to identify the major manifestations. The follow-up of these patients should be extended, due to the polymorphic nature of the syndrome. Care should be taken when performing surgical procedures or with prolonged immobilization, due to the predisposition for deep vein thrombosis. Psychosocial and educational follow-up should be carried out8,9. The surgeries will consist of the resection of large tumors with abundant vascularization. Conventional and ultrasonic liposuction were tried in our first two cases, aiming to reduce fatty tumors without damaging vessels. Both methods were insufficient, and provided few aesthetic or functional benefits for the patients. Both subsequently underwent surgical resection of the tumors, which were easily accessible, without the abundant bleeding expected, and with good outcomes. It is important to have blood products available before starting a procedure, due to the potential risk of bleeding.
REFERENCES
1. Cohen MM Jr, Hayden PW. A newly recognized hamartomatous syndrome. Birth Defects Orig Artic Ser. 1979;15(5B):291-6. PMID: 118782
2. Wiedemann HR, Burgio GR, Aldenhoff P, Kunze J, Kaufmann HJ, Schirg E. The proteus syndrome. Partial gigantism of the hands and/or feet, nevi, hemihypertrophy, subcutaneous tumors, macrocephaly or other skull anomalies and possible accelerated growth and visceral affections. Eur J Pediatr. 1983;140(1):5-12. PMID: 6873112
3. Happle R. Lethal genes surviving by mosaicism: a possible explanation for sporadic birth defects involving the skin. J Am Acad Dermatol. 1987;16(4):899-906. PMID: 3033033 DOI: http://dx.doi.org/10.1016/S0190-9622(87)80249-9
4. Wiedemann HR, Burgio GR. Encephalocraniocutaneous lipomatosis and Proteus syndrome. Am J Med Genet. 1986;25(2):403-4. PMID: 3777031
5. Cohen MM Jr. Further diagnostic thoughts about the Elephant Man. Am J Med Genet. 1988;29(4):777-82.
6. Stricker S. Musculoskeletal manifestations of Proteus syndrome: report of two cases with literature review. J Pediatr Orthop. 1992;12(5):667-74.
7. Miura H, Uchida Y, Ihara K, Sugioka Y. Macrodactyly in Proteus syndrome. J Hand Surg Br. 1993;18(3):308-9. PMID: 8345255 DOI: http://dx.doi.org/10.1016/0266-7681(93)90047-J
8. Biesecker LG, Happle R, Mulliken JB, Weksberg R, Graham JM Jr, Viljoen DL, et al. Proteus syndrome: diagnostic criteria, differential diagnosis, and patient evaluation. Am J Med Genet. 1999;84(5):389-95. PMID: 10360391 DOI: http://dx.doi.org/10.1002/(SICI)1096-8628(19990611)84:5<389::AID-AJMG1>3.0.CO;2-O
9. Cohen MM Jr. Proteus syndrome: an update. Am J Med Genet C Semin Med Genet. 2005;137C(1):38-52. PMID: 16010681 DOI: http://dx.doi.org/10.1002/ajmg.c.30063
10. Villela ALC, Guedes LGS, Paschoa VVA, David AB, Tenório TM, Lamego Junior HP, et al. Perfil epidemiológico de 58 portadores de síndrome de Klippel-Trénaunay-Weber acompanhados no Ambulatório da Santa Casa de São Paulo. J Vasc Bras. 2009;8(3):219-24.
11. Clark RD, Donnai D, Rogers J, Cooper J, Baraitser M. Proteus syndrome: an expanded phenotype. Am J Med Genet. 1987;27(1):99-117. PMID: 3605210 DOI: http://dx.doi.org/10.1002/ajmg.1320270111
12. Cohen MM Jr, Turner JT, Biesecker LG. Proteus syndrome: misdiagnosis with PTEN mutations. Am J Med Genet A. 2003;122A(4):323-4. PMID: 14518070
13. Gordon PL, Wilroy RS, Lasater OE, Cohen MM Jr. Neoplasms in Proteus syndrome. Am J Med Genet. 1995;57(1):74-8. PMID: 7645604 DOI: http://dx.doi.org/10.1002/ajmg.1320570117
14. Jamis-Dow CA, Turner J, Biesecker LG, Choyke PL. Radiologic manifestations of Proteus syndrome. Radiographics. 2004;24(4):1051-68. DOI: http://dx.doi.org/10.1148/rg.244035726
Universidade Federal do Paraná, Curitiba, PR, Brazil
Institution: Disciplina de Cirurgia Plástica da Universidade Federal do Paraná, Curitiba, PR, Brazil.
Corresponding author:
Renato da Silva Freitas
Rua General Carneiro, 191
Curitiba, PR, Brazil Zip code 80860-900
E-mail: dr.renato.freitas@gmail.com
Article received: December 24, 2011.
Article accepted: February 24, 2012.
 Read in Portuguese
Read in Portuguese
 Read in English
Read in English
 PDF PT
PDF PT
 Print
Print
 Send this article by email
Send this article by email
 How to Cite
How to Cite
 Mendeley
Mendeley
 Pocket
Pocket