


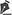
Case Report - Year 2014 - Volume 29 -
Infantile digital fibromatosis: case report
Fibromatose digital infantil: relato de caso
ABSTRACT
INTRODUCTION: Infantile digital fibromatosis, also known as Reye tumor, is a rare, asymptomatic, benign nodular proliferation of fibrous tissue, which occurs almost exclusively in the dorsolateral region of the fingers and toes. This article reports a case of infantile digital fibromatosis.
CASE REPORT: This case was diagnosed by clinical, imaging, and histopathological examination. The patient presented at the Orthopedic Department of our hospital, with a 4-year history of a painless, erythematous nodular lesion on the second toe of the left foot. On physical examination, a deformity of the second toe caused by a nodular, erythematous, painless lesion of approximately 1.5 cm diameter was noted; the lesion did not result in functional changes. Ultrasound examination revealed a solid, hypoechoic nodule involving the extensor tendon in the middle phalanx of the second toe. The initial diagnosis was fibroma or synovioma. Due to the clinical characteristics of the lesion, its evolution, and the imaging findings, the team chose to perform a biopsy. However, due to the small size of the lesion, upon open biopsy, surgical excision was performed. Histopathological examination confirmed the diagnosis of infantile digital fibromatosis.
CONCLUSION: Infantile digital fibromatosis is a rare clinical entity, which should be differentiated from other lesions found in the fingers and toes. The correct diagnosis is rarely made pre-operatively, due mainly to a failure to recognize this entity. For this reason, it is essential to onsider this lesion in the differential diagnosis of digital nodules.
Keywords: Foot; Infantile digital fibromatosis; Reye tumor; Case Report.
RESUMO
INTRODUÇÃO: A fibromatose digital infantil é uma proliferação nodular, assintomática, rara e benigna do tecido fibroso, que ocorre quase exclusivamente na região dorsal e lateral dos dedos das mãos e pés. O artigo relata um caso de fibromatose digital infantil, também conhecida como tumor de Reye.
RELATO DE CASO: Trata-se de um caso diagnosticado por meio de exames clínico, de imagem e histopatológico. O paciente apresentou-se ao Setor de Ortopedia do Hospital, queixando-se de uma lesão nodular, eritematosa, indolor, no segundo pododáctilo do pé esquerdo, existente havia quatro anos. Durante o exame físico, notava-se uma deformidade no II pododáctilo, causada por uma lesão nodular, eritematosa, indolor, de aproximadamente 1,5cm, que não acarretava alterações funcionais. O exame de ultrassom revelou a presença de uma imagem nodular sólida, hipoecogênica, envolvendo o tendão do extensor do II pododáctilo na falange média. O diagnóstico inicial era de fibroma ou sinovioma. Pelas características clínicas da lesão, por seu tempo de evolução e pelos achados de imagem, a equipe optou por uma biópsia. No entanto, devido ao pequeno tamanho da lesão, sendo a biópsia aberta, realizou-se a exérese cirúrgica. O exame histopatológico confirmou o diagnóstico de fibromatose digital infantil.
CONCLUSÃO: Esse tumor constitui uma entidade clínica rara, que deve ser diferenciada de outras lesões encontradas nos dedos das mãos e dos pés. O diagnóstico correto raramente é feito antes da operação, devido, principalmente, à falha em reconhecer essa entidade. Por essa razão, é essencial considerar essa lesão em diagnósticos diferenciais.
Palavras-chave: Pé; Fibromatose digital infantil; Tumor de Reye; Relato de caso.
Fibromatosis was first described by Stout, and was an important advance in the study of soft tissue tumors 1. Fibromatoses encompass a wide spectrum of benign proliferations of fibrous tissue, with similar histological findings and biological behavior, intermediate between that of benign and malignant lesions. These neoplasms are unencapsulated tumors, able to infiltrate adjacent structures without forming regional or systemic metastases. Currently, the most widely accepted and used system of classification is that proposed by Enzinger and Weiss, in which lesions are classified as either a superficial fibromatosis (fascial) or a deep fibromatosis (musculoaponeurotic)2.
Superficial fibromatoses, which tend to be small and slow-growing, include palmar and plantar fibromatosis, juvenile aponeurotic fibroma, and infantile digital fibromatosis. Deep fibromatoses are large and can grow rapidly; they are more aggressive, with a tendency to grow from the deep fascia, over the muscle and the aponeurotic tissue. Examples of deep fibromatoses include infantile myofibromatosis, fibromatosis colli, extra-abdominal desmoid tumor, and aggressive infantile fibromatosis.
Infantile digital fibromatosis is a rare and benign lesion, most commonly found in the fingers and toes, which mainly affects the dorsal and lateral region 3-13. The lesion was first described by Reye, in 1965, as a recurring digital fibrous tumor 7. According to Werther and Seiersen (1997), approximately 100 cases of this condition have been reported in the literature 3. Despite these 100 cases, the literature offers no consensus on the ideal treatment of this condition 1-13.
The etiology of infantile digital fibromatosis is unknown; there is no sex difference in prevalence, and no familial tendency. In one third of cases, the lesion is congenital 3. The condition is little-known, even in orthopedic and dermatological specialties, likely due to its rarity. It is, however, essential to consider this condition in the differential diagnosis of lesions of the feet and hands in pediatric patients, in order to avoid misguided procedures. After evolving over 2-3 years, these lesions tend to regress in approximately 12% of patients; however, approximately 60% of surgical cases experience a recurrence 3,4,10,12-13.
This report describes a case of infantile digital fibromatosis in the second toe of the left foot of a 5-year-old patient; the diagnosis was reached by a combination of clinical, imaging, and histopathological examination.
CASE REPORT
The patient, a 5-year-old male child with leucoderma, presented to the Orthopedics Department of our hospital on 28 August 2004, with the complaint of a nodular lesion in the second toe of the left foot. There was no clinical history of previous trauma, comorbidities, or other relevant findings that could aid in the diagnosis. On physical examination, a painless, erythematous nodular lesion, approximately 1.5cm diameter, was observed in the dorsolateral region of the left second toe, between the middle and the distal phalanx (Figure 1). The lesion did not result in any impairment of function.
Figure 1. Patient with a nodular lesion, approximately 1.5 cm diameter, on the dorsolateral region of the second toe of the left foot.
Ultrasound examination revealed a hypoechoic solid nodule that involved the extensor tendon of the second left toe, at the middle phalanx; the nodule measured 12x8.2x4.7 mm in longitudinal, transverse, and anteroposterior diameter, respectively. A diagnosis of fibroma or synovioma was suspected.
Based on the clinical characteristics of the lesion, its evolution over a period of approximately 4 years, and the imaging findings, the team chose to perform a biopsy under local anesthetic block with sedation. However, due to the small size of the lesion, upon open biopsy a surgical excision was performed, with margins of + -3mm. Primary closure of the lesion was achieved with a simple suture.
Macroscopic histopathological examination revealed an ellipse of skin measuring 1.5x0.5x0.4cm, with a raised area on the skin surface. Incisions at this level showed a firm whitish tissue. Accompanying tissue fragments also were firm and whitish, with overall dimensions of 1.5x1.5x0.5cm. Microscopic examination revealed histological sections of skin fragment showing the preserved epidermis. There was a proliferation of fibroblasts in the dermis, surrounded by a dense collagen stroma. The fibroblasts were arranged in bundles that extended from the deep layer of the epidermis to the dermis, forming a poorly-defined nodule. Masson's trichrome staining revealed small intracytoplasmic and eosinophilic inclusions, close to the nucleus of the fibroblasts (Figure 2). The pathologist concluded the report stating that the findings were consistent with infantile digital fibromatosis.
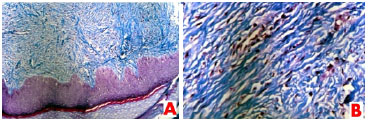
Figure 2. Photomicrograph of a section of the excised lesion. (A); Histological slide revealing preserved epidermal tissue with diffuse fibroblast proliferation in the dermis. Masson's trichrome staining (100x). (B); Slide showing small eosinophilic intracytoplasmic inclusion bodies, located close to the nuclei of the fibroblasts. Masson's trichrome staining (400x).
After surgery, the patient was instructed to return to the orthopedic department for observation. At a follow-up appointment in February 2005, approximately 5 months after surgery, a growth with the same clinical features as the original lesion was observed. Ultrasound imaging was requested on 15 February 2005; the examination showed a hypoechoic solid nodule, involving the extensor tendon of the second left toe at the middle phalanx. The patient was diagnosed with recurrent infantile digital fibromatosis, and expectant management was chosen; this involved educating the family about the benign nature of the condition, and the probability of spontaneous regression.
DISCUSSION
Infantile digital fibromatosis, also known as Reye tumor, is a rare and benign tumor. This fibroma differs from other forms of fibroma in 3 aspects: it is limited to the digits of pediatric patients; it shows a remarkable tendency to relapse, reportedly in over 60% of cases; and histopathological examination shows intracytoplasmic inclusions 3,4, 6,7. The lesion was first described by Reye, in 1965, as a recurring digital fibrous tumor 5.
The etiology of infantile digital fibromatosis is unknown. A viral etiology was initially suspected, in view of the presence of intracytoplasmic inclusion bodies found on histopathological examination; however the results of electron microscopy and polymerase chain reaction failed to support this hypothesis 9.
From an embryonic perspective, the mesoderm gives rise to mesoblasts, and, thereafter, to pre-fibroblasts and fibroblasts. The pre-fibroblast, depending on influencing factors, can differentiate into a wide variety of connective tissue cells, including fibroblasts, myoblasts, and lipoblasts; the pre-fibroblast can also differentiate into other kinds of cells, including osteoblasts, chondroblasts, blood cells, and lymphoblasts. The fibroblast deserves particular attention because it has a high oncogenic potential and may lead to a variety of tumors ranging from benign infantile digital fibromatosis to the deadly aggressive fibromatosis; other tumors, including malignant fibrous histiocytoma, infantile myofibromatosis, desmoid tumor, and fibrosarcoma, fall between these extremes9.
There are multiple factors that can affect the oncogenic potential of fibroblasts, including genetic, sex-linked, hormonal, and growth-related factors. Although not confirmed, this is suggested, in the case of infantile digital fibromatosis, by the presence of intracytoplasmic inclusion bodies. It has also been proposed that the circulation of abnormal levels of maternal hormones in newborn blood results in the rapid appearance of tumors shortly after birth, and explains their subsequent spontaneous involution 9.
The nodules of infantile digital fibromatosis may be single or multiple, firm or gelatinous in consistency, and erythematous. Nodules may reach over 2cm in diameter, and develop almost exclusively in the fingers and toes, particularly in the dorsolateral region; in most cases the thumb and the first toe are unaffected 3-5. There have been rare reports of similar lesions with intracytoplasmic inclusion bodies in extra-digital sites, including the arms, feet, breasts, and tongue 3-5.
One third of lesions are congenital, and the majority of nodules appear within the first months of life, while approximately 75% of cases are identified in the first year of life1-8. Reports of infantile digital fibromatosis in older children or adults are extremely rare. There is no sex difference in prevalence, no familial tendency, and no association with a prior history of trauma. The diagnosis accounts for 0.1% of fibrous tumors in children 5,8,10.
Differential diagnosis is performed with relative ease, on the basis of the location, age of subjects, and the presence of intracytoplasmic inclusion bodies, a unique feature of infantile digital fibromatosis. Differential diagnoses consist of acquired digital fibrokeratoma, dermatofibroma, annular granuloma, neurilemoma, sarcoidosis, angiofibroma, and fibrosarcoma 4,5,8,10,11. A biopsy is recommended in all cases to confirm the diagnosis 4,5,11,13.
The histopathological findings in infantile digital fibromatosis are unique. The fascicles of interdigitating sheets of elongated cells and collagen bundles in the dermis forms the node 9, which can extend up to the subcutaneous tissue. The oval nuclei of the cells are accompanied by intracytoplasmic inclusion bodies that correspond with actin microfilaments. The epidermis over the lesion can present hyperkeratosis or acanthosis. The adnexal structures and the fat tissue may be surrounded by proliferating fibrous tissue. Unusually, this tissue may extend to the periosteum and cause erosion, although the bone itself is never affected. Immunohistochemistry is positive for vimentin, cytokeratin, desmin and muscle-specific actin, suggestive of a myofibroblastic origin. Immunohistochemistry is negative for S-100 protein and glial fibrillary acidic protein, and is reported to be positive for CD57 in some cases 9.
With regard to treatment, in view of the benign pathology, more recent studies recommend conservative management4,7. In approximately 12% of reported cases there was spontaneous regression in an average of 2-3 years3,4,6,8. Surgery is only recommended in cases of deformity or functional change3,4,6-8. According to Dabney et al., in a review of 92 reported cases, 78 were treated surgically; recurrence occurred in 64% and 10% required amputation10. Mohs micrographic surgery may be used in cases with functional impairment, although the rate of recurrence remains high11,12. Corticosteroids and topical imiquimod showed no benefit, although corticosteroid infiltration of the lesion may be beneficial. The major drug used is triamcinolone at a dose of 10mg/mL intralesionally; depending on the lesion diameter, this is equivalent to a total of 0.1-0.3mL/lesion. The dose can be repeated within 4-6 weeks until adequate lesion regression is observed3,4. There are also reports of successful infiltration with fluorouracil13.
The main complications of infantile digital fibromatosis include deformity, functional changes and, in some cases, ulceration4. These complications are rare, and the overall prognosis is excellent; in 12% of cases, there is spontaneous regression of the lesion in 2-3 years, and there are no reports of malignant change or metastasis in the literature. However, a notable feature of this pathology is the high recurrence rate, estimated to be in the region of 60% 3-5,11-13.
CONCLUSION
Infantile digital fibromatosis is a rare clinical entity that should be differentiated from other causes of nodular lesions presenting on the fingers and toes. The correct diagnosis is rarely made prior to surgery, mainly due to a failure to recognize this entity. Consequently it is essential to consider this lesion in the differential diagnosis, in order to avoid misguided procedures. We stress the importance of educating family members about the benign nature of the lesion.
REFERENCES
1. Stout AP. Juvenile fibromatoses. Cancer. 1954;7(5):953-78.
2. Enzinger FM, Weiss SW. "Fibromatosis", in Soft tissue tumors, 3rd ed., St. Louis, C.V. Mosby, 1995;201-229.
3. Werther K, Seiersen M. Recurrent infantile digital fibromatosis. Ugeskr Laeger. 1997;159:4656-7.
4. Ishii N, Matsui K, Ichiyama S, Takahashi Y, Nakajima H. A case of infantile digital fibromatosis showing spontaneous regression. Br J Dermatol. 1989;121:129-33.
5. Reye RD. Recurring digital fibrous tumors of childhood. Arch Pathol. 1965;80:228-31.
6. Kawaguchi M, Mitsuhashi Y, Hozumi Y, Kondo S. A case of infantile digital fibromatosis with spontaneous regression. J Dermatol. 1998;25:523-6.
7. De León BB, Fernández VJ. Fibromatosis con cuerpos de inclusión. An Med Assoc Med Hosp ABC. 2004;49(3):147-50.
8. Baser NT, Tuncali D, Akbuga UB, Aslan G. Infantile digital fibromatosis: late results of two different treatment approaches. Eur J Plast Surg. 2006;29:38-40.
9. Zhu WY, Xia MY, Huang YF, Leonardi C, Penneys NS. Infantile digital fibromatosis: ultrastructural human papillomavirus and herpes simplex virus DNA observation. Pediatr Dermatol. 1991;8:137-9.
10. Dabney KW, MacEwen GD, Davis NE. Recurring digital fibrous tumor of childhood: case report with long-term follow-up and review of the literature. J Pediatr Orthop. 1986;6(5):612-7.
11. Albertini JG, Welsch MJ, Conger LA, et al. Infantile digital fibroma treated with Mohs micrographic surgery. Dermatol Surg. 2002;28:959-61.
12. Campbell LB, Petrick MG. Mohs micrographic surgery for a problematic infantile digital fibroma. Dermatol Surg. 2007;33:385-7.
13. Oh CK, Son HS, Kwon YW, Jang HS, Kwon KS. Intralesional fluorouracil injection in infantile digital fibromatosis. Arch Dermatol. 2005;141:549-50.
1 - Orthopedist and Traumatologist specializing in Hand Surgery. Member of the Brazilian Society of Hand Surgery. Orthopedist and Traumatologist, hand surgery specialist at Hospital Geral Governador Israel Pinheiro - IPSEMG - of Belo Horizonte and Hospital Unimed Betim/MG
2 - Plastic surgeon, specialist member of the Brazilian Society of Plastic Surgery (BSPS), clinical staff member of Unimed Hospital, Biocor Institute, Hospital Vera Cruz and the Institute of Mastology, Odontology and Plastic Surgery (IMOP), Belo Horizonte, MG, Brazil
3 - Orthopedist and Traumatologist, Hospital Unimed Betim/MG. Specialist in Knee Arthroscopic Surgery. Orthopedist and Traumatologist, Hospital Unimed Betim/MG
Institution: Hospital Unimed Betim.
Corresponding author:
Gustavo Augusto Matos Saliba
Avenida Governador Valadares, nº 454, Centro
Betim, MG, Brasil
E-mail: salibagustavo@yahoo.com.br
Article received: May 23, 2011
Article accepted: July 29, 2011
 Read in Portuguese
Read in Portuguese
 Read in English
Read in English
 PDF PT
PDF PT
 Print
Print
 Send this article by email
Send this article by email
 How to Cite
How to Cite
 Mendeley
Mendeley
 Pocket
Pocket