ISSN Online: 2177-1235 | ISSN Print: 1983-5175


Body and Chest - Year2011 - Volume26 - (3 Suppl.1)
Relatar um caso de neurofibroma abdominal gigante, discutindo seus aspectos clínicos e patológicos e demonstrar o tratamento cirúrgico empregado.
RELATO DO CASO
J.G.S., 14 anos, sexo feminino,raça negra, proveniente do estado da Bahia, radicada em São José do Rio Preto há 2 meses. Queixando-se de "excesso de pele em abdome" congêita, com aumento progressivo anual, negando dor à palpação ou trauma local. Paciente sem comorbidades, referindo caso semelhante na família - pai e irmão portadores da mesma patologia, também de forma congêita, porém com menor intensidade. Relata anemia crônica tratada com sulfato ferroso. Ao exame físico, observa-se aumento localizado de tecido subcutâneo em flanco direito de aproximadamente 20 cm de diâmetro e hipogástrio direito de aproximadamente 7 cm de diâmetro. Presença de hipercromia da epiderme, com aumento da rugosidade e flacidez local. Após anamnese e exame físico detalhados, acrescidos das fotos realizadas, a paciente submeteu-se a exames de imagem, tais como ultra-sonografia e tomografia computadorizada de parede abdominal, os quais revelaram em detalhes a localização e a extensão do tumor. A cirurgia foi realizada sob anestesia geral, pela equipe de cirurgia plástica do Hospital Santa Casa de Misericórdia de São José do Rio Preto. Optado por ressecção em fuso, obedecendo linha de dobra da própria lesão, semelhante a abdominoplastia clássica. Realizado pontos de adesão entre o subcutâneo e aponeurose, na tentativa de melhor fixação do retalho e reduzir a chance de complicações como seroma. Deixado dreno a vácuo até o segundo pós-operatório, recebendo alta hospitalar. O peso da peça foi aproximadamente 3,0 kg.
DISCUSSÃO
A Neurofibromatose tipo I (NF-1) é também conhecida como síndrome de von Recklinghausen. Compreende, juntamente com a neurofibromatose tipo II, a esclerose tuberosa, a síndrome de Sturge-Weber e a síndrome de von Hippel-Lindau, o conjunto de doenças conhecidas como facomatoses (ou síndromes neurocutâneos). Afeta primariamente o crescimento das células dos tecidos neurais, determinando o aparecimento de tumores benignos que se instalam nos locais mais variados e que podem surgir em qualquer etapa da vida. A doença é conhecida há séculos, datando de 1592 a primeira descrição de um portador provável e, de 1768, o primeiro caso confirmado publicado. Em 1882, von Recklinghausen descreveu a patologia de maneira correta e praticamente completa, confirmando então a origem nervosa dos tumores. A neurofibromatose tipo 1 é uma das anomalias autossômicas dominantes mais frequentes na espécie humana, a pessoa portadora de neurofibromatose tem um risco de 50% de ter um filho também com neurofibromatose, risco este considerado alto. O gene NF1 está localizado no cromossomo 17 na posição 17q11.2. Critérios propostos pelo NIH (1988): 1. Seis ou mais manchas café com leite com diâmetro maior que 1,5 cm em adolescentes ou adultos ou 0,5 cm em crianças; 2. Dois ou mais neurofibromas de qualquer tipo ou apenas um neurofibroma plexiforme; 3. Dois ou mais nódulos de Lisch (esferas acastanhadas da íris importantes para o diagnóstico, não trazendo nenhuma diminuição visual a seus portadores).
CONCLUSÃO
Nos casos de neurofibromas plexiformes gigantes em tronco e membros, após completa investigação diagnóstica por métodos clínicos e de imagem, a simples ressecção cirúrgica e reconstrução plástica mostrou-se um método eficaz e satisfatório de correção do problema, tanto para a equipe médica quanto para a paciente.
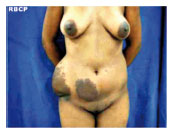
Figura 1 - Pré-operatório, vista anterior.
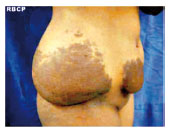
Figura 2 - Pré-operatório, vista lateral direita.
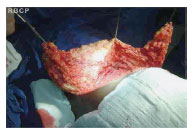
Figura 3 - Intraoperatório, confecção do retalho abdominal.
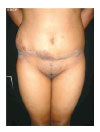
Figura 4 - Pós-operatório, vista anterior.
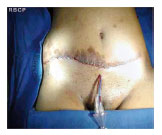
Figura 5 - Pós-operatório imediato.
Indexers

 All scientific articles published at www.rbcp.org.br are licensed under a Creative Commons license
All scientific articles published at www.rbcp.org.br are licensed under a Creative Commons license